Hit Identification Service
Virtual Screening
We offer in silico screening services for any protein-target with a solved 3D structure on the basis of our 100 million compounds library. Our high-quality screening services command a hit rate of 10-50%. This means that half of the molecules calculated (or predicted) for the experimental activity assessment are genuinely potent inhibitors. Our screening team much more often successfully applies proprietary drug discovery software because it uses laws of quantum and molecular physics instead of the widespread but poor quality statistical approaches based on scoring functions and QSAR-like methods.
Compound profiling prediction
The compound profile is predicted by virtual screening of a drug candidate molecule against hundreds of diverse proteins representing different active site types of a human proteome. Compound profiling provides a list of comprehensive prediction effects: we identify potential targets and predict therapeutic and adverse effects because we screen a compound in silico against diverse protein sets representing the human proteome.
As a result we obtain Kd of the tested compound for every protein from our carefully preselected protein set. Active site types that show high affinity for the compound are considered as sensitive. On the second round of screening the tested compound is screened against all biologically important proteins with sensitive active site types and solved 3D structures.
Optionally, the screening can be run against specialized protein assays such as kinase assays. As opposed to the compound profiling in vitro/vivo, in silico compound profiling does not require synthesis and/or purchasing of chemicals. In addition, determination of cumulative effects takes from several months to several years, while virtual compound profiling does this within several hours or days. Finally, compound profiling in silico saves the lives of many animals.
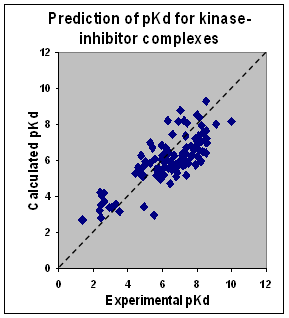
Kinase Profiling assays — a unique option in silico platform!
Kinase profiling identifies prospective safe, effective and selective kinase inhibitors when we virtually screen a compound against our kinase profiling platform. Kinase profiling in silico does not require complicated and expensive synthesis of drug candidates and acquiring of kinase assays and kits. Our process cuts out associated time delays.
Our kinase assays represent practically all major kinase groups of the human kinome. The table below shows the number of kinase families from each group represented in our kinase platform.
 |
|
We constantly revise and expand assays with newly-solved kinase structures.
Compound profiling procedure
Accuracy
Aurora's compound profiling protein set
Virtual High Throughput Screening
Our virtual screening identifies prospective strong inhibitors for a protein target as a customer indicates. We conduct hit identification by docking huge virtual structure libraries on a target protein.
Screening in silico dramatically decreases the number of compounds for experimental assessment of their activity (cutting down time and financial expenses) and increases the percentage success of in vitro experiments. Virtual screening is an effective way to rationalize drug-design. We know that Aurora's virtual high throughput screening can successfully substitute HTS and UHTS for protein-targets with solved 3D structures.
The hit identification procedure consists of four steps. First, we divide the library into clusters according to structural similarity of compounds. Second, we dock the representatives of every cluster on a rigid structure model of a protein-target. Third, we dock the whole clusters containing identified hits on a rigid structure model of a protein-target. Fourth, we send the best docked molecules, with IC50 < 10um to a refinement calculation. Our refinement procedure is a complete Free Energy Perturbation of molecular dynamics run for a whole protein-ligand complex in aqueous environment, which takes into account protein flexibility.
Our hit rate for predicted strong inhibitors is from 10% to 50%.
Chemical and physiological properties prediction
This service provides essential information on the vulnerability of compounds to help direct medicinal chemistry activities and pharmacokinetic modeling.
The following values and properties can be calculated and predicted using our software:
Solubility in Water/DMSO prediction
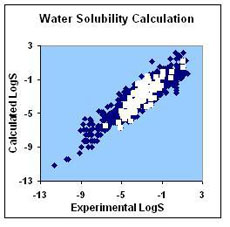
The main advantage of Aurora's software is the quality of the underlying physical models. Most competing approaches use different kinds of fragment based descriptors to calculate the molecular properties from known properties of similar compounds (QSAR). Such models that rely heavily on additivity of molecular properties are often overparametrized and lack direct physical justification. As a result, the prediction power of the models may be very good for structures similar to those used in the training set and may not be sufficient for absolutely novel compounds. Aurora's software derives molecular properties from first principles based on direct models using advanced quantum mechanical analysis of molecular interactions and thermodynamics. The service predicts both water and dimethylsulfoxide (DMSO) solubility of organic compounds at various temperatures and pH values (from 0.0 to 14.0). The accuracy of calculations for most structures is usually better than 0.2-0.5 logS units; for more complicated molecules the error can be up to 0.8-1.1 logS units. Such parameters as the solvent temperature, pH and ion strength are adjustable. The results of the calculations are represented both in logarithmic (LogS) and absolute (g/l) units.
The figures below demonstrate calculated solubility in water and DMSO for over 1300 and 60 compounds, correspondingly, plotted against their experimental values. The white points on the water solubility graph represent the LogS values calculated for a number of commercially available drugs taken from the Drug Bank database. The blue points show our performance for a set of generic molecules taken from the Virtual Computational Chemistry Lab. The points on the DMSO solubility graph represent the LogS values calculated for a number of chemicals taken from the Gaylord Chemical Corporation database.
 |
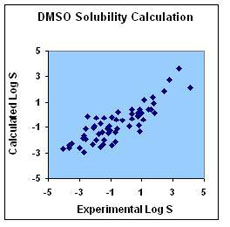 |
| RMSD = 0.81 LogS units; R2 = 0.84 | RMSD = 0.70 LogS units; R2 = 0.72 |
The compound structures can be supplied in one of the commonly used chemical file formats such as .hin, .pdb, .sdf, .mol2. The final report includes aqueous and DMSO solubility in logarithmic and absolute units (g/l). The results of the calculations can be saved in Excel-readable (.csv) format.
LogP and LogD Calculation
LogP - partition coefficient between lipid membranes and water
The main advantage of Aurora's software is the quality of the underlying physical models. Most of the competing approaches use different kinds of fragment based descriptors to calculate the molecular properties from known properties of similar compounds (QSAR). Such models that rely heavily on the additivity of molecular properties are often overparametrized and lack direct physical justification. As a result, the prediction power of the models may be very good for structures similar to those used in the training set and may not be sufficient for absolutely novel compounds. Aurora�'s software derives the molecular properties from first principles based on direct models using advanced quantum mechanical analysis of molecular interactions and thermodynamics.
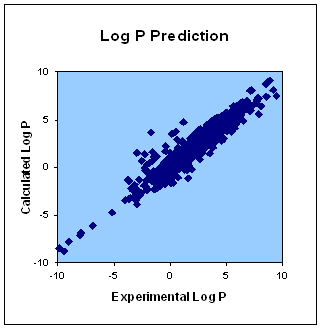
The service predicts water/octanol partition coefficient, LogP, for any low weight organic molecule (both charged and non-charged ones), calculates LogD for dissociative systems at a given pH and drug-likeness according to Lipinski's rule. Such parameters as the solvent temperature and ionic strength are adjustable.
Mean square deviation between calculated and experimental values is 0.7 Log P units. That is of the same order of accuracy as the currently used experimental techniques for determining LogP!
The figure demonstrates calculated Log P values plotted against their experimental ones for over 900 organic molecules (actually drugs from The Drug Bank database). The calculation showed excellent correlation with experimental values: R2 = 0.94.
The Ligands can be supplied in one of the commonly used chemical file formats such as .hin, .pdb, .sdf, .mol2. For LogD calculation pKa value is necessary. If its experimental value is unavailable, order our pKa calculation service first to predict these values and then to calculate LogD.
The output data includes LogP and LogD in logarithmic units and estimation of compliance of the drugs with Lipinski's rule of 5:
Plasma protein binding - affinity constant for serum albumin;
pKa - the negative log to base 10 of the acid dissociation constant;
LogBB - blood-brain barrier permeation;
Lipinski's Rule of Five, an evaluation of druglikeness.
Virtual Identification of a Protein Target
This service identifies a target protein for active compounds (e.g., compounds from natural substrates: herbs, venoms, etc). If possessing or having developed an active compound, there is an interest in the identification of the protein target which the compound is working against, we can offer a fast and cost efficient identification method for the desired target.
The identification process includes virtual screening of a compound against a few representative proteins. The active sites of the proteins with high affinity to the compound are considered as sensitive. The compound will be screened against all proteins containing certain sensitive active sites. As a result of this procedure the desired protein will be identified.
The same process can be applied to a complex mixture containing many compounds such as, for example, natural extracts. One does not need to separate and isolate every mixture ingredient.
It will be enough to proceed to the virtual screening for every component as shown above and to find out the active compound and the appropriate desired protein.